Veille Bibliographique pour le site GFRS – Résumé par Brigitte GRANEL – Avril 2026
Scleromyositis – The new frontier
HudsonM et al. Best Pract Res Clin Rheumatol. 2026 Mar 6:102126.
Introduction et objectif(s) de l’étude
L’attente musculaire est peu connue au cours de la Sclérodermie Systémique (ScS), or elle est responsable d’une importante morbidité. Cet article présente les caractéristiques cliniques-sérologiques-histopathologiques de l’atteinte musculaire de la ScS et propose le terme de scléromyosite (SM), considérant l’atteinte musculaire propre à la ScS et non plus comme un syndrome de chevauchement.
Méthode(s)
Les auteurs s’attachent à la définition de la SM : elle ne se limite pas à la découverte d’anticorps anti-PM/Scl. La SM est décrite comme une association entre les critères de ScS et ceux d’une myosite incluant des critères cliniques (faiblesse musculaire, myalgies), biologiques avec créatine kinase (CK) supérieure à 5N, électroneuromyographiques ou histologiques à la biopsie musculaire. Cependant, les critères histopathologiques classiquement utilisés dans les myosites n’incluent pas certaines caractéristiques importantes de la ScS, telles que la fibrose et la vasculopathie. Ainsi, le spectre de la SM semble beaucoup plus large que ce qui a été reconnu jusqu’à présent, en utilisant l’association des critères de classification de la ScS à ceux des myosites ou les simples anticorps anti-PM/Scl.
Résultat(s)
Une étude sur 3919 patients a montré que 672 (17%) présentaient une myosite, définie comme un antécédent de myopathie déclaré par le médecin et/ou faiblesse proximale avec au moins un test objectif anormal tel qu’une CK élevée, un tracé de souffrance musculaire à l’électromyogramme, un œdème musculaire à l’IRM ou une histologie positive. Les auto-anticorps les plus courants étaient les anticorps anti-PM/Scl, -U3RNP et -Ku, tandis que l’anticorps anti-centromères était protecteur. La forme cutanée diffuse de la ScS était plus fréquente et la myopathie était associée à un risque accru de décès.
L’analyse de la littérature montre que les patients ne remplissent pas toujours les critères de classification cliniques et sérologiques pour la ScS et que d’autres caractéristiques qui ne sont pas dans les critères de ScS, en particulier l’arthrite, l’atteinte gastro-intestinale, la calcinose et l’atteinte cardiaque sont utiles pour étayer un diagnostic de SM. De même, l’élévation de la CK n’est pas la règle car des SM peuvent avoir un taux de CK normal. A l’imagerie par résonance magnétique (IRM), les anomalies sont fréquentes : œdème musculaire, atrophie, infiltration graisseuse et fibrose, sans corrélation entre la faiblesse musculaire et le taux de CK. Il est également important de noter que près de la moitié des patients atteints de SM n’avaient pas d’auto-anticorps connus (SM « séronégatives »). Les principaux auto-anticorps courants et leurs spécificités sont résumés dans la Figure. Les caractéristiques histopathologiques de la SM sont hétérogènes : inflammation lymphocytaire, nécrose ou fibrose, et surtout anomalies capillaires. Cette atteinte microvasculaire, caractéristique pour les auteurs, soutient le concept de SM comme une manifestation organique de la ScS plutôt qu’un chevauchement avec une autre maladie musculaire. Ainsi, une évaluation capillaire systématique est essentielle et peut être réalisée par immunomarquage du CD31 ou encore plus efficacement avec un marqueur de membrane basale tel le collagène-4 qui peut démontrer à la fois l’élargissement capillaire et le relâchement, et un indice de la réplication de la membrane basale des capillaires.
Conclusion
La prise en charge repose essentiellement sur des données observatoires et le niveau de preuve est faible en l’absence de recommandations publiées spécifiquement sur le sujet. Le principe reste de minimiser l’exposition aux corticoïdes (< 15 mg/j) à cause du risque de crise rénale sclérodermique, et l’adaptation des traitements immunosuppresseurs en fonction des autres organes atteints. Cependant, des doses plus élevées de cortisone peuvent être justifiées dans la SM, en particulier dans les cas d’atteinte musculaire sévère, ou d’atteinte diaphragmatique. Le méthotrexate ou le mycophénolate mofétil sont proposés, le Rituximab peut être associé dans les formes les plus sévères. Les immunoglobulines intraveineuses peuvent être utiles en épargne cortisonique, ou chez les patients présentant une atteinte musculaire sévère, incluant dysphagie, faiblesse diaphragmatique et axiale, ou chez ceux avec une myocardite. Les mesures associés (renutrition, activité physique) sont impératives.
Message(s) clé
-L’atteinte musculaire de la ScS est encore sous diagnostiquée, mise à tort sous le compte de nombreux facteurs comme la dénutrition, la sclérose cutanée, l’atteinte articulaire, cardiaque, pulmonaire ou un simple déconditionnement. Elle peut être la première manifestation de la ScS, alors que les autres manifestations ne sont pas encore présentes, ce qui en retarde le diagnostic. -La biopsie musculaire est un outil à la fois de diagnostic et d’aide dans la prise en charge thérapeutique.
-Les auteurs proposent la vasculopathie comme point d’entrée unique expliquant l’atteinte musculaire de la SM.
-La SM récapitule la triade pathologique : vasculopathie, inflammation et fibrose.
-Comme pour d’autres manifestations de la ScS, une reconnaissance et une intervention précoce minimiseront les complications et les lésions irréversibles.
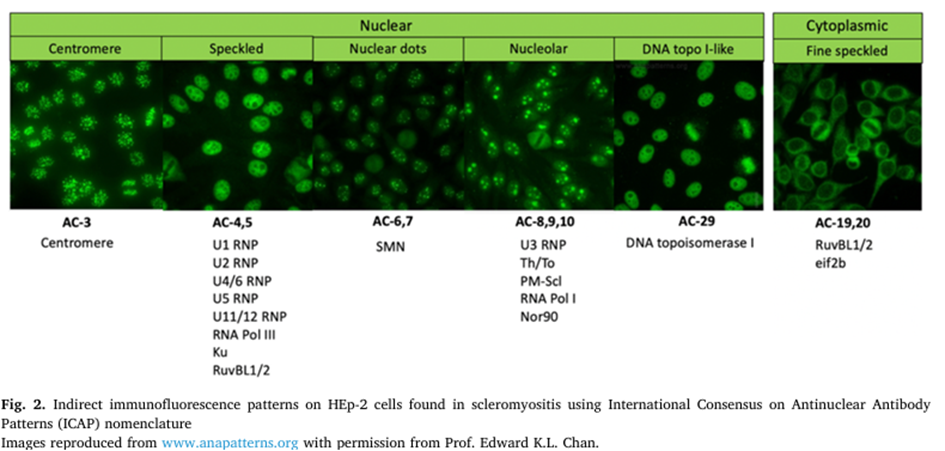
Figure issue intégralement de l’article.
Veille Bibliographique pour le site GFRS – Résumé par Brigitte GRANEL – Avril 2026
Hudson M, Troyanov Y, Landon-Cardinal O. Scleromyositis – The new frontier
Best Pract Res Clin Rheumatol 2026 Mar 6:102126. doi: 10.1016/j.berh.2026.102126.