Poumon et Sclérodermie Systémique. Lung and Systemic Sclerosis.
Mise au point. Auteur : Éric Hachulla.
Rev Med Interne. 2026; 47(2):70-75. https://doi.org/10.1016/j.revmed.2025.08.006
Introduction et objectif(s) de l’étude
La pneumopathie interstitielle diffuse (PID) est une atteinte fréquente de la sclérodermie systémique (ScS), touchant 40 à 50% des patients (jusqu’à 70 à 100% sur les données autopsiques), grevant le pronostic. La PID et l’hypertension artérielle pulmonaire sont les 2 atteintes pulmonaires prédominantes et les 2 principales causes de décès de cette maladie.
Méthode(s)
Il s’agit d’une mise au point concise basée sur la revue de la littérature dont l’objectif est de faire le point sur le dépistage, les facteurs de facteurs de risques et la thérapeutique actuelle.
Résultat(s)
Le dépistage systématique de la dyspnée et la réalisation d’un scanner thoracique haute résolution et d’épreuves fonctionnelles respiratoires (EFR) avec capacité de diffusion pulmonaire (DLCO) sont recommandés dès le diagnostic. Ces 3 mesures sont importantes, car environ un tiers des patients avec ScS et PID n’ont pas de dyspnée à l’effort et la moitié des patients avec ScS et des EFR normaux ont une PID.
Le suivi EFR avec DLCO et la clinique est recommandé tous les 6 mois durant les 3 à 5 premières années. Un nouveau scanner doit être réalisé pour évaluer la progression si les EFR ou la DLCO s’aggravent. Certains patients sont plus à risque de développer une PID et seront à surveiller de façon plus assidue (Tableau 1).
L’aspect de « pneumopathie interstitielle non spécifique (PINS) » est le plus fréquent (80%). La « pneumopathie interstitielle commune (PIC) » est plus rare (10%). La PID est étendue si ≥ 30 % de fibrose au CT-scan, ou 10 à 30% de fibrose avec une capacité vitale forcée (CVF) < 70% du prédit. La PID est limitée si ≤ 10 % de fibrose au CT-scan, ou 10 à 30% de fibrose avec une CVF ≥ 70 % du prédit (critères de Goh).
Le syndrome emphysème des sommets et fibrose des bases, observé exclusivement chez le fumeur, est une forme grave.
Au plan thérapeutique, les recommandations actuelles du Protocole national de diagnostic et de soins
(PNDS, Figure 1), de l’American Thoracic Society, l’European Alliance of Associations for Rheumatology (EULAR) préconisent une prise en charge personnalisée basée sur le risque évolutif.
Le mycophénolate mofétil (MMF, hors AMM) est le traitement oral de première ligne. Le rituximab (RTX, hors AMM) a montré un effet favorable sur la stabilisation voire l’amélioration de la CVF. Le MMF et le rituximab ont été positionnés en première ligne dans l’atteinte de la PID-Sclérodermique dans les recommandations de l’EULAR 2023. Le tocilizumab (hors AMM) a également démontré un ralentissement du déclin de la CVF dans les sous-groupes avec PID dans deux essais randomisés. Le nintédanib (AMM) a prouvé son efficacité dans l’essai SENSCIS, réduisant significativement le déclin annuel de la CVF, y compris chez les patients recevant du MMF. En cas de progression malgré un traitement initial bien conduit, une autogreffe de cellules souches hématopoïétique ou une combinaison MMF + RTX peut être discutée. La transplantation pulmonaire peut être envisagée dans les formes réfractaires. Une approche intégrative associant oxygénothérapie, réhabilitation et prophylaxie infectieuse est essentielle.
Conclusion
Il est important de dépister précocement la PID de la ScS, d’en assurer le suivi et une prise en charge thérapeutique médicamenteuse selon sa gravité initiale ou sa progression.
Message(s) clé
Il est important de dépister, suivre et traiter la PID de la ScS. Une discussion thérapeutique est nécessaire dès son diagnostic et la thérapie à valider de façon collégiale, le plus souvent en RCP.

Tableau issue intégralement de l’article.
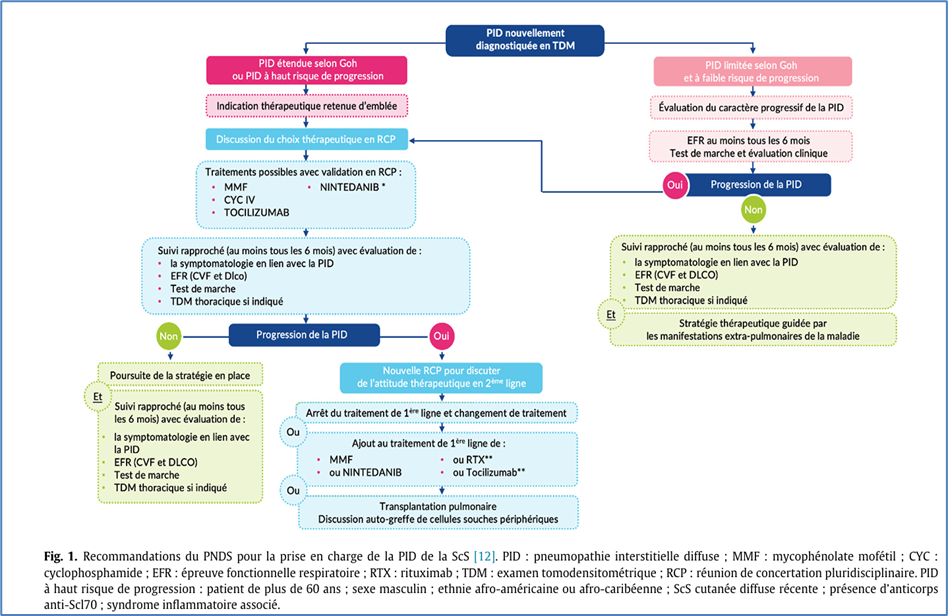
Figure 1 : algorithme des recommandations générales du PNDS publié en 2022 (https://www.has-sante.fr/upload/docs/application/pdf/2008-11/pnds__sclerodermie_web.pdf).
(Figure issue intégralement de l’article).
Hachulla E. Poumon et sclérodermie systémique. Rev Méd Interne 2026 ; 47(2):70-75.